
1.2. ПАТОГЕНЕЗ
Патогенез ИЭ довольно сложен и окончательно не изучен. Принципиальную схему
патогенеза ИЭ можно представить следующим образом: врождённые, приобрётенные
дефекты клапанов сердца увеличение скорости и появление турбулентности
трансклапанного потока крови механическое повреждение эндотелия клапанов
отложение тромбоцитов и фибрина на повреждённых участках эндокарда формирование
хронического неинфекционного эндокардита с тромботическими вегетациями
преходящая бактериемия на фоне снижения реактивности организма адгезия и
колонизация патогенных бактерий в фибрино-тромбоцитарных вегетациях воспаление
эндокарда, формирование микробных вегетаций, разрушение клапанов развитие СН,
системного инфекционного процесса с эмболическим, тромбогеморрагическим,
иммунокомплексным поражением внутренних органов и тканей (рисунок 1).
В качестве инициальных механизмов патогенеза выделяют повреждение эндокарда,
бактериемию, адгезию, размножение, колонизацию патогенных бактерий на клапанах.
Основная роль в развитии ИЭ принадлежит деструкции эндокарда, бактериемии.
Экспериментальные исследования свидетельствуют о том, что катетеризация сердца в
течение нескольких минут вызывает чувствительность эндокарда к микробной
агрессии на протяжении многих дней.
Данные электронной микроскопии позволили проследить последовательность
формирования патологического процесса. Выяснено, что под воздействием
регургитирующего потока крови изменяется форма и структура эндотелиоцитов,
увеличивается межклеточная проницаемость, происходит десквамация эндотелия.
Между эндотелиоцитами образуются поры, через которые проникают лимфоциты,
макрофаги. Увеличение размеров пор, снижение атромбогенных свойств эндокарда
усиливает адгезию бактерий. На месте отслоения дистрофически изменённых клеток
идёт интенсивное тромбообразование. Эндокард покрывается активированными
тромбоцитами, “прошивается” волокнами фибрина.
Повреждение, деэндотелизация эндокарда усиливают адгезию бактерий,
образование покрывающего слоя из тромбоцитов, фибрина. Создается недоступная для
фагоцитов “зона локального агранулоцитоза”, обеспечивающая выживание,
размножение патогенных микроорганизмов. В процессе продолжающейся бактериальной
колонизации, нарастания тромбоцитарно-фибринового матрикса формируются микробные
тромбы, вегетации, происходит повреждение, разрушение клапана.
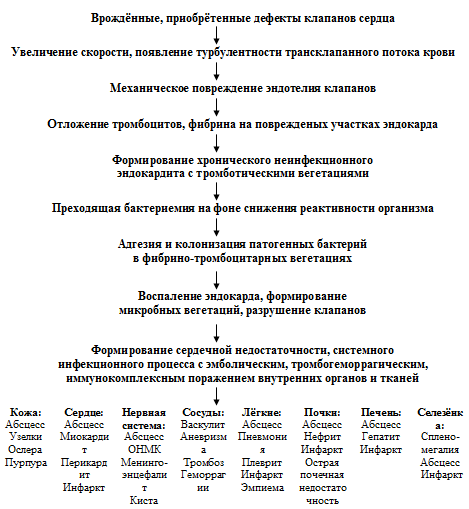
Рисунок 1. Схема патогенеза ИЭ (Демин А.А., Дробышева В.П.,
2005).
Факторы, усиливающие адгезию бактерий к эндокарду, можно условно разделить на
местные и общие. В состав местных входят врождённые и приобретённые изменения
клапанов, нарушение внутрисердечной гемодинамики. Врождённые пороки увеличивают
риск трансформации бактериемии в ИЭ до 92%. Предрасполагающие условия для
возникновения заболевания создают механические, биологические искусственные
клапаны. К общим факторам относят нарушения резистентности организма, выраженные
изменения иммунитета, развивающиеся при проведении иммунносупрессивной терапии,
у наркоманов, больных алкоголизмом, людей пожилого возраста и пациентов, имеющих
изменения в HLA-системе гистосовместимости.
Формирование ИЭ происходит на фоне бактериемии, травмы эндокарда, снижения
резистентности организма. Бактериемии принадлежит ведущая роль. Источниками
бактериемии могут быть очаги хронической инфекции, инвазивные медицинские
исследования и манипуляции (бронхоскопия, гастроскопия, колоноскопия,
хирургические вмешательства), тонзилэктомия, аденоидэктомия, вскрытие и
дренирование инфицированных тканей, стоматологические процедуры.
Развитие ИЭ зависит от массивности, частоты, видовой специфичности
бактериемии. Риск развития заболевания особенно велик при повторяющихся
“минимальных” или “массивной” бактериемии вследствие хирургических операций.
Бактериемия staph. aureus является стопроцентным фактором риска ИЭ в связи
повышенной адгезией и связыванием пептидогликаном эндокарда этих бактерий.
Значительно меньшая вирулентность у эпидермального стафилококка и стрептококков.
Вероятность развития ИЭ при пневмококковой бактериемии составляет приблизительно
30 %.
Существуют определённые закономерности в локализации инфекции, обусловленные
нарушением внутрисердечной гемодинамики при формировании порока. Такими
анатомическими образованиями при недостаточности клапанов являются поверхность
МК со стороны левого предсердия, поверхность АК со стороны аорты, хорды. При
незаращении межжелудочковой перегородки чаще поражается эндокард правого
желудочка в области дефекта.
Персистирующая бактериемия стимулирует иммунитет, запуская
иммунопатологические механизмы воспаления. Изменения иммунитета при ИЭ
проявляются гипофункцией Т-лимфоцитов, гиперфункцией В-лимфоцитов, поликлоновой
продукцией аутоантител. Нарушаются механизмы активации комплемента, образуются
циркулирующие иммунные комплексы. В современных исследованиях подтверждается
значительная патогенетическая роль увеличения концентрации ЦИК с отложением в
органах-мишенях. Несомненного внимания заслуживает увеличение концентрации
интерлейкинов 1, 6, 8 и фактора некроза опухоли, провоспалительная активность
которых наряду с индукцией острофазового ответа, участвует в развитии системных
проявлений ИЭ.
Тромбоэмболии способствуют генерализации инфекционного процесса, формированию
инфарктов, некроза органов. У 52-67 % больных ИЭ с преимущественным поражением
правых камер сердца развивается тромбоэмболия лёгочной артерии. Обструкция
сосуда сопровождается гуморальными нарушениями, возникающими в результате
выброса биологически активных веществ из агрегатов тромбоцитов в тромбе (тромбоксан,
гистамин, серотонин).
При ТЭЛА образуются “мёртвые” пространства в лёгких (несколько сегментов или
доля), не перфузируемые смешанной венозной кровью. Значительно возрастает
шунтирование смешанной венозной крови в лёгких. Снижение градиента напряжения
углекислого газа между смешанной венозной и артериальной кровью, повышение
концентрации углекислого газа в артериальной крови вызывает артериальную
гипоксемию.
Увеличение общего сосудистого лёгочного сопротивления кровотоку - один из
основных механизмов формирования артериальной лёгочной гипертензии у больных ИЭ.
Изменения гемодинамики и реологии крови вызывают неадекватную перфузию
сосудистых зон, расстройство газообмена. Снижение доставки кислорода к лёгочной
ткани, накопление тканевых метаболитов и токсичных продуктов анаэробных
процессов являются причиной развития инфаркта лёгкого.
В развитии хронической СН у больных ИЭ выделяют несколько патогенетических
механизмов: формирование недостаточности клапана (ов), септическое поражение
миокарда, перикарда, изменения гемодинамики, нарушение ритма, проводимости,
задержка жидкости, связанная с нарушением функции почек. Важным звеном
патогенеза СН является увеличение постнагрузки при длительном повышении
периферического сосудистого сопротивления. Вазоконстрикция обусловливает
поддержание системного артериального давления, оптимизирует сниженный сердечный
выброс.
Недостаточность МК вызывает дилатацию, гипертрофию левых отделов сердца
повышение давления в сосудах малого круга кровообращения декомпенсацию по
левожелудочковому типу гипертрофию правого желудочка сердечную недостаточность
по большому кругу. Повреждение аортального клапана способствует развитию
диастолической перегрузки левого желудочка гипертрофии, дилатации левого
желудочка относительной недостаточности МК (“митрализация порока”) гипертрофии,
дилатации левого предсердия застою крови в малом круге кровообращения,
декомпенсации по левожелудочковому типу гипертрофии, дилатации правых отделов
сердца правожелудочковой СН. Выраженная недостаточность трёхстворчатого клапана
вызывает дилатацию, гипертрофию правого предсердия дилатацию, гипертрофию
правого желудочка вследствие поступления в его полость увеличенного объёма крови
из правого предсердия венозный застой в большом круге кровообращения.
При ИЭ изменяются микроциркуляция, реологические свойства крови. Происходит
внутрисосудистое свёртывание крови, которое в своём развитии проходит четыре
стадии. Первая стадия гиперкоагуляции и компенсаторного гиперфибринолиза
начинается в поражённом органе, из клеток высвобождаются коагуляционно-активные
вещества, активация коагуляции распространяется на кровь. Вторая стадия
нарастающей коагулопатии потребления и непостоянной фибринолитической активности
характеризуется уменьшением числа тромбоцитов, концентрации фибриногена в крови.
Третья стадия дефибриногенации и тотального, но не постоянного фибринолиза (дефибриногенационно-фибринолитическая),
соответствует полному ДВС-синдрому. Четвёртая - стадия остаточных тромбозов и
окклюзий.
Причинами нарушения микроциркуляции являются микротромбозы, ремоделирование
микрососудов. Изменение геометрии сосудов начинается как адаптивный процесс при
нарушении гемодинамики, повышении активности тканевых, гуморальных факторов. В
последующем ремоделирование сосудов способствует прогрессированию расстройств
кровообращения. Изменения микроциркуляции обусловлены повышенной агрегацией
тромбоцитов, эритроцитов. При левожелудочковой СН на фоне периваскулярного отёка
происходит агрегация эритроцитов, локальный эритростаз, фрагментация кровотока.
Особая роль отводится повышенной активности плазменного гемостаза. Значение
гиперфибриногенемии, как самостоятельного фактора снижения реологических свойств
крови и прогрессирования ИЭ, обосновано в клинико-экспериментальных
исследованиях. Важное значение в нарушении микрогемодинамики имеет образование
микротромбов. Гемореологические изменения вызывают снижение перфузионных свойств
крови, усиливают расстройства гемодинамики на периферии. Нарастает тканевая
гипоксия, активизируется аэробный метаболизм. Гипоксия тканей при хронической СН
снижает сократительную способность миокарда, увеличивает пред- и постнагрузку.
В течении ИЭ различают несколько патогенетических фаз:
инфекционно-токсическую (септическую), иммуновоспалительную, дистрофическую.
Первая фаза характеризуется транзиторной бактериемией с адгезией патогенных
бактерий на эндотелий, формированием микробнотромботических вегетаций. Вторая
фаза проявляется полиорганной патологией (эндоваскулит, миокардит, перикардит,
гепатит, нефрит, диффузный гломерулонефрит).
Под воздействием эндогенных токсинов происходит декомпенсация органов и
систем, нарушается метаболизм, происходит дезинтеграция организма как
биологического целого. Во время дистрофической фазы формируются тяжёлые,
необратимые изменения внутренних органов.
Указанные патогенетические фазы типичны для всех клинико-морфологических форм и
вариантов течения болезни. Тем не менее, патогенез вторичного ИЭ имеет некоторые
особенности. Врождённый порок сердца увеличивает функциональную нагрузку на
сердечно-сосудистую систему и клапаны, повреждается эндотелий. Функция органов,
богатых ретикулоэндотелиальной тканью, подвержена угнетению. Снижается
неспецифическая резистентность организма. Преходящая бактериемия вызывает
образование первичного инфекционного очага.
На фоне снижения общей резистентности формируется хронический воспалительный
процесс. Развивается сенсибилизация организма бактериальными антигенами. Миокард
повреждается кардиальными антителами. Во время бактериемии из очагов хронической
инфекции происходит адгезия бактерий на изменённых клапанах. Формируется
вторичный септический очаг в сердце, являющийся основой для развития вторичного
ИЭ.
Инфекционный эндокардит с поражением правых камер сердца развивается после
повреждения ТК подключичным катетером, при зондировании сердца, продолжительном
стоянии катетера Swan-Ganz, выполнении частых внутривенных инъекций. Широкое
применение катетеризации сосудов с целью интенсивной инфузионной терапии
увеличивает количество случаев тромбофлебита, тромбоза, инфицирования с
последующим развитием сепсиса.
Следует отметить, что 30 % катетеров подключичной вены достигают полости
правого предсердия сердца и травмирует створки ТК. Установка эндокардиальных
электродов для электрокардиостимуляции в ряде случаев является причиной
инфекционного поражения ТК. Причиной развития ИЭ в правых камерах сердца могут
быть пули, осколки других огнестрельных снарядов, длительно находящиеся в
сердце.
Вторичный ИЭ с поражением правых камер сердца чаще развивается при дефекте
межжелудочковой перегородки, открытом артериальном протоке (22%). Развитие ИЭ
обусловлено повреждением эндокарда регургитирующим потоком крови. При высоких
небольших дефектах межжелудочковой перегородки тонкая струя крови травмирует
септальную створку ТК. В случае открытого артериального протока травмируется
эндокардиальная поверхность лёгочного ствола в области дефекта. Таким образом, в
последние десятилетия наиболее частой причиной развития первичного ИЭ является
сеспсис, внутривенная наркомания, а вторичного - врождённые пороки сердца.
Для развития ИЭ у наркоманов типично повреждение эндокарда при частых
внутривенных инъекциях. Во время инъекций самостоятельно изготовленных
наркотических веществ пузырьки воздуха повреждают эндокард трёхстворчатого
клапана в 100 % случаев. Травмируется эндокард, возникает его шероховатость.
Повреждённые участки служат местом адгезии, агрегации тромбоцитов с последующим
формированием тромбов. Нарушение асептики способствует развитию бактериемии,
инфицированию повреждённых участков эндокарда золотистым стафилококком (70-80
%). Причина его тропности к эндокарду ТК у наркоманов не совсем ясна.
Изменения иммунитета, неспецифической резистентности являются ключевыми
механизмами патогенеза этой формы болезани. По данным исследования иммунного
статуса у больных ИЭ с поражением правых камер сердца выявлено уменьшение
Т-хелперов, увеличение Т-супрессоров, снижение активности натуральных киллеров.
Эти изменения вызваны угнетением реактивности иммунной системы вследствие
истощения функциональных резервов. Зарегистрировано увеличение концентрации ФНО
- цитокина, играющего ключевую роль в развитии иммунно-воспалительных реакций
организма.
Среди многочисленных эффектов ФНО обращает на себя внимание его действие на
коллаген клапанов 1, 3, 4-го типов, составляющий 50-70 % его массы. Фактор
некроза опухоли ингибирует транскрипцию гена коллагена, тем самым, снижая синтез
последнего фибробластами. Кроме того, ФНО стимулирует продукцию коллагеназы,
которая участвует в деградации коллагена клапанов. Денатурированные фрагменты
коллагена индуцируют продукцию воспалительных медиаторов макрофагами, индуцируют
и поддерживают воспалительный процесс.
Число наркоманов и пациентов, у которых длительно используются катетеры
сосудов велико. Однако, не у всех развивается ИЭ. В этой связи изучены
генетические аспекты предрасположенности. По данным исследования HLA-фенотипа
(по антигенам локусов А, В) наиболее вероятными маркерами генетической
предрасположенности к ИЭ с поражением правых камер сердца являются антиген
системы HLA В35, гаплотип А2-В35.
Структурной основой изменения реактивности иммунной системы у больных являются
нарушения пространственной организации комплекса: Т-клеточный рецептор -
иммуногенный пептид - белок главного комплекса гистосовместимости. В развитии
заболевания имеет значение сочетание генетической детерминированности дефекта
иммунной системы с модификацией антигенов гистосовместимости инфекционными
агентами, химическими веществами (наркотики, антибиотики) и другими факторами.
Развитие ИЭ протеза клапана обусловлено многими причинами: травматизацией
эндокарда во время операции, бактериемией, снижением резистентности организма,
изменением иммунитета. При протезировании искусственных клапанов происходит
инфицирование, которое определяется физическими свойствами, химическим составом
имплантируемого клапана, адгезией бактерий на шовном материале. Повышенная
адгезия стафилококков на внутрисердечных швах определяет состав возбудителей
раннего ИЭПК (staphylococcus epidermidis, staphylococcus aureus).
В 50 % случаев раннего ИЭПК источником бактериемии является послеоперационная
рана. В патогенезе позднего ИЭПК ключевое значение имеет транзиторная
бактериемия, возникающая при интеркуррентных инфекциях (36%), стоматологических
манипуляциях (24 %), операциях (12 %), урологических исследованиях (8 %).
Дополнительными источниками инфекции являются артериальные системы,
внутривенные, уретральные катетеры, сердечные заплаты, эндотрахеальные трубки.
Инфекция начинается с абактериальных тромботических наложений, которые затем
инфицируются при транзиторной бактериемии. Большие гемодинамические нагрузки
являются причиной развития ИЭ искусственного клапана, находящегося в митральной
позиции. Воспаление начинается с манжеты протеза, фиброзного кольца. Далее
образуются аннулярные, кольцевые абсцессы, формируются парапротезные фистулы,
происходит отрыв протеза.
Таким образом, развитие инфекционного эндокардита обусловлено
иммунодефицитом, первичным или вторичным повреждением эндокарда, приходящей
бактериемией. Дальнейшее течение болезни опосредовано комплексом
патогенетических механизмов, которые формируются в результате системного
поражения сосудов, множественных тромбоэмболий, иммунокомплексных реакций,
изменения центральной и внутрисердечной гемодинамики, нарушения свёртывающей
системы крови. |