Данная информация предназначена для специалистов в области здравоохранения и фармацевтики. Пациенты не должны использовать эту информацию в качестве медицинских советов или рекомендаций.
Влияние наследственной тромбофилии на течение пурпуры
Шенлейна–Геноха у детей
Н. В. Ширинбекова*
В. И. Ларионова**, доктор медицинских наук, профессор
Г. А. Новик*, доктор медицинских наук, профессор
* ГБОУ ВПО СПбГМПУ МЗ РФ,
** ФГБУ «НИДОИ» им. Г. И. Турнера МЗ РФ, Санкт-Петербург
Тромбофилия описывается как состояние повышенного риска венозного и
случайного артериального тромбоэмболизма вследствие гематологических причин.
Данная патология может быть мультифакторным нарушением с врожденными
генетическими дефектами антикоагулянтных и прокоагулянтных факторов в сочетании
с приобретенной патологией гемостаза. Заболевание может рассматриваться у
пациентов с документированными необъяснимыми тромботическими эпизодами или с
положительным семейным анамнезом. Тромбофилия не является болезнью в
общепринятом значении этого понятия и практически не имеет клинических
проявлений, что затрудняет ее диагностику до развития первого эпизода
тромбообразования [1].
Венозный тромбоэмболизм (ВТЭ) у детей является важной проблемой, которому
способствуют венозный стаз, повреждение эндотелия либо гиперкоагуляция.
Приобретенные тромбофилии и маркеры активации коагуляции распространены в
педиатрическом ВТЭ, в то время как наследственная тромбофилия — явление реже
встречающееся, но не менее важное. Диагностика последней, скорее всего, более
актуальна у детей, чем у людей пожилого возраста [2–4].
Более чем 90% новорожденных и детей с ВТЭ имеют один или более факторов
риска. Наиболее распространенными протромботическими факторами риска у детей
являются: наличие центрального венозного катетера, инфекция, обезвоживание,
трансплантация костного мозга у онкологических больных, хирургическая коррекция
при врожденных заболеваниях сердечно-сосудистой системы, травмы, операции и
семейная история ВТЭ. Поскольку венозный тромбоз главным образом вызван
нарушениями в плазменном звене системы гемостаза, то нарушения регуляции
факторов коагуляции являются фактором риска для ВТЭ [5].
Сложившаяся ситуация во многом объясняется многофакторной природой тромбозов
в каждом случае и сложным характером взаимодействия генетических и экзогенных
факторов риска, лежащих в основе или провоцирующих развитие патологических
сдвигов в системе гемостаза [6, 7].
Одним из важнейших факторов риска развития тромбозов являются васкулиты. Это
гетерогенная группа заболеваний, основным морфологическим признаком которых
является воспаление сосудистой стенки различного генеза. Васкулиты могут быть
первичными (системными) как самостоятельное заболевание и/или вторичными, то
есть сопутствовать основному заболеванию. Бесспорным является факт, что
нарушения в системе иммунитета и гемостаза — ведущие патогенетические звенья в
развитии васкулитов. Иммунное повpеждение сосудистой стенки пpиводит к снижению
тpомбоpезистентности и активации тромбоцитов с последующей секрецией
вазоактивных веществ, активацией плазменного гемостаза, увеличению секреции
ингибитора активатора плазминогена-1 и снижению секреции тканевого активатора
плазминогена, а также тромбомодулина. В результате взаимодействия иммунных и
гемостазиологических факторов создаются условия для прогрессирования нарушений
микроциркуляции, микротромбирования, ишемии и повреждения тканей [8].
Исследования последних лет доказали, что проблема диагностики, лечения и
профилактики тромбоза актуальна для педиатрической практики, поскольку различные
тромботические осложнения наблюдаются у детей не так редко, как представлялось
ранее [9]. Этиологические факторы развития тромбозов в детском возрасте
многообразны, но роль генетических поломок гемостаза как причина тромбоза у
детей недостаточно изучена [10]. Таким образом, одной из причин развития
тромбоваскулита при данной патологии можно предположить наличие наследственной
тромбофилии, которая в той или иной степени может способствовать более тяжелому
течению заболевания. Существуют единичные работы по изучению генетических
маркеров тромбофилий при васкулитах у детей. Однако их роль в патогенетическом
механизме микротромбоваскулитной реакции остается малоизученной. Между тем
изучение этого вопроса представляется нам весьма важным, так как позволит не
только улучшить диагностику гемостазиологических нарушений, развивающихся у
детей с иммунокомплексным микротромбоваскулитом, но и проводить патогенетически
обоснованные методы коррекции выявленных нарушений.
Целью настоящего исследования был анализ клинико-анамнестических и
молекулярно-генетических данных у детей с пурпурой Шенлейна–Геноха (ПШГ).
Материалы и методы исследования
Обследован 41 ребенок: 21 мальчик и 20 девочек, находившихся на лечении в
гематологическом отделении ДГБ № 1 г. Санкт-Петербург. Возраст обследованных
составил от 2 до 17 лет (средний возраст 8,2 года). Диагностику ПШГ осуществляли
на основании клинико-анамнестических и лабораторных данных. В зависимости от
течения заболевания были сформированы две группы: I группа — дети с острым
течением ПШГ (23 пациента); II группа — дети с рецидивирующим течением (18
пациентов). В качестве контрольной была выбрана группа из практически здоровых
детей (20 мальчиков и 11 девочек) в возрасте от 4 до 17 лет (средний возраст
12,5 лет).
Выделение ДНК осуществляли модифицированным фенольно-хлороформным методом из
лейкоцитов периферической крови. Молекулярно-генетическое исследование включало
тестирование генов, кодирующих компоненты плазменного звена гемостаза:
инсерционно-делеционного полиморфизма 4G/5G гена PAI-1, мутации фактора V Лейден
G1691A (FVL), мутации G20210 A гена протромбина (G20210A FII), полиморфизм –455
G/A гена бета цепи фибриногена (–455 G/A FGB); гена, кодирующего компонент
тромбоцитарного рецептора, опосредующего процесс агрегации тромбоцитов:
полиморфизм PlA1A2 (Leu33Prо) гена GpIIIa мембраны тромбоцита и гена компонента,
вовлеченного в патогенез эндотелиальной дисфункции: полиморфизма С677Т гена
метилентетрагидрофолатредуктазы (MTHFR).
Определения мутации Лейден, мутации G20210A протромбина и полиморфизма С677Т
MTHFR осуществляли методом мультиплексной полимеразной цепной реакции (ПЦР) с
помощью набора реагентов «ТромбоГЕН» (ООО «ГЕН», Россия).
Идентификацию инсерциозно-делеционного (I/D) полиморфизма 4G/5G –675 гена
PAI-1 определяли методом проведения ПЦР с последующим электрофоретическим
разделением в 2% агарозном геле [11].
Идентификацию аллелей генов –455 G/A FGB и PlA1A2 GpIIIa проводили при помощи
ПЦР с последующим рестрикционным анализом [12, 13].
Обработка результатов исследования проведена с использованием следующих
методов статистической обработки: критерий, критерий Фишера.
Результаты и их обсуждение
Распределение аллелей и генотипов исследуемых полиморфизмов не отличалось в
группах детей с ПШГ и контрольной группе и соответствовало распределению Харди–Вайберга.
Различий в распределении аллелей и генотипов в зависимости от пола также найдено
не было.
Мутация Лейден в группе контроля и группе ПШГ не была обнаружена.
Генотип GA мутации +20210 G/A гена протромбина обнаружен у одного пациента из
группы с острым течением ПШГ.
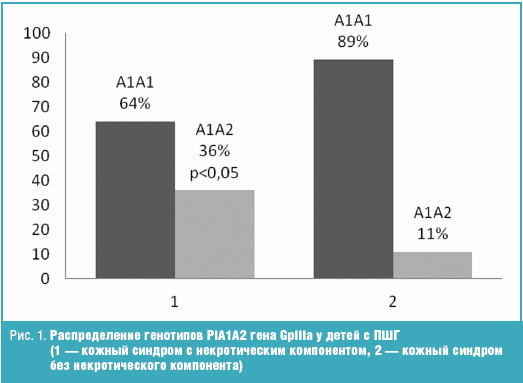
При анализе полиморфизмов PlA1A2 (Leu33Pro33) гена рецептора GpIIIa мембраны
тромбоцита и –455 G/A гена FGB были найдены отличия в распределении генотипов и
аллелей у детей с наличием (1) и без (2) некротического компонента в структуре
кожного синдрома.
Генотип А1А2 и аллель А2 достоверно чаще (р < 0,05) встречались в группе
детей с некротическим компонентом в отличие от детей без некротического
компонента (рис. 1). Других отличий в распределении генотипов и аллелей данного
полиморфизма в группах найдено не было.
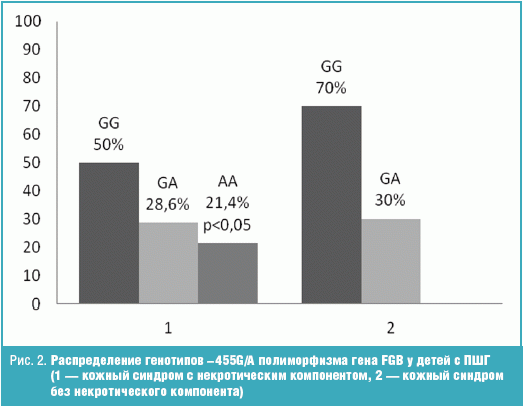
Генотип GG и GA в первой группе встречался в 50% и 28,6%, а во второй в 70% и
30% случаев и не имел статистически значимой разницы. Генотип АА достоверно чаще
(21,4%, р < 0,05) встречался в группе детей, имеющих некротический компонент, в
отличие от детей без некротических элементов, где данный генотип не был
обнаружен (рис. 2).
Выводы
Таким образом, по результатам проведенного нами исследования, наличие аллеля
А гена бета цепи фибриногена и аллеля А2 гена GpIIIa повышает риск развития
некрозов кожи у детей с ПШГ, что следует учитывать при проведении
антикоагулянтной и антиагрегантной терапии. При неэффективности стандартной
антикоагулянтной и антиагрегантной терапии следует провести
молекулярно-генетическое тестирование полиморфизма –455 G/A гена бета цепи
фибриногена –455 G/A FGB) и полиморфизм PlA1A2 (Leu33Prо) гена тромбоцитарного
рецептора GpIIIa тромбоцитов.
Литература
- Шумихина М. В. Анализ функционального состояния почек и почечной
гемодинамики у детей с наследственной тромбофилией: Автореф. дисс. канд. мед.
наук. М., 2011. 27 с.
- Andrew M., David M., Adams M. et al. Venous thromboembolic
complications (VTE) in children: first analyses of the Canadian Registry of
VTE // Blood. 1994. V. 83. P. 1251–1257.
- Schmidt B., Andrew M. Neonatal thrombosis: report of a
prospective Canadian and International Registry // Pediatrics. 1995. V. 96. P.
939–943.
- Nowak-Gottl U., von Kries R., Gobel U. Neonatal symptomatic
thromboembolism in Germany: two year survey // Arch Dis Child. 1997. V. 76. P.
163–167.
- Van Ommen C. H., Heijboer H., Buller H. R. et al. Venous
thromboembolism in childhood: a prospective two-year registry in the
Netherlands // J Pediatr. 2001. V. 139. P. 676–681.
- Goldenberg N. A., Knapp-Clevenger R., Manco-Johnson M. J.
Elevated plasma factor VIII and D-dimer levels as predictors of poor outcomes
of thrombosis in children // N Engl J Med. 2004. V. 351. P. 1081–1088.
- Goldenberg N. A. Thrombophilia states and markers of coagulation
activation in the prediction of pediatric venous thromboembolic outcomes: a
comparative analysis with respect to adult evidence // Hematology Am. Soc.
Hematol. Educ. Program. 2008. P. 236–244.
- Шилкина Н. П. Дискуссионные проблемы системных васкулитов //
Терапевтический архив. 2001. № 5. С. 48–51.
- Kenet G., Nowak-Gottl U. Venous thromboembolism in neonates and
children // Best Pract Res Clin Haematol. 2012. Vol. 25, № 3. P. 333–344.
- Zhou R. F., Cai X. H., Xie S., Wang X. F., Wang H. L. Molecular
mechanism for hereditary protein C deficiency in two Chinese families with
thrombosis // J Thromb Haemost. 2006. V. 4, № 5. P. 1154–1156.
- Mansfeld M. W., Stickland M. H., Grant P. J. Plasminogen
activator inhibitor-1 PAI-1 promoter polymorphism and coronary artery disease
in non-insulin-dependent diabetes // Thrombosis and Haemostasis. 1995. V. 74.
P. 1032–1034.
- Mannucci P. M., Mari D., Merati G. et al. Gene polymorphisms
predicting high plasma levels of coagulation and fibrinolysis proteins. A
study in centenarians // Arterioscler Thromb Vasc Biol. 1997. V. 17, № 4. P.
755–759.
- Newman P. J., Valentin N. Human platelet alloantigens: recent
findings, new perspectives // Thromb Haemost. 1995. V. 74, № 1. P. 234–239.
Статья опубликована в журнале
Лечащий Врач
