
2.5. Этиология и патогенетические механизмы
Патогенетические механизмы дистоний пока остаются раскрытыми лишь
частично. Дистония не имеет четкого морфологического субстрата в мозге и
обусловлена субклеточными и нейродинамическими нарушениями в определенных
мозговых системах. Известно, что периферический моторный аппарат, пирамидный
путь, проприоцептивный серво-механизм (стреч-рефлекс) интактны при дистонии.
Выявлены нарушения в функциональном состоянии интернейронов ствола головного
мозга и спинного мозга. При проведении радиологических нейровизуализационных
исследований при первичных дистониях не обнаружены как структурные изменения,
так и врожденные нарушения метаболизма (Голик В.А., Марченко С.В., 2007). Тем не
менее, широта вовлечения в патогенез дистонии отдельных образований нервной
системы представляется неограниченной (рис. 1).
Почти неизвестен биохимический дефект, лежащий в основе дистонии.
Эмпирически можно предполагать заинтересованность холинергических,
дофаминергических и ГАМКергических систем мозга. Но низкая эффективность лечения
дистоний в целом предполагает существование каких-то иных, нам еще неизвестных
биохимических нарушений, лежащих в основе заболевания. Скорее всего, триггером,
запускающим дистонию, являются биохимические системы орального отдела ствола
головного мозга и его связей с подкорковыми экстрапирамидными образованиями
(главным образом скорлупой, зрительным бугром и другими).
К настоящему времени утвердилось представление о том, что
патологический процесс при гиперкинезах не столько очерчен какой-то более или
менее грубой локальной анатомической деструкцией, сколько представляет собой
сложный патофункциональный комплекс, развивающийся по законам нейродинамики с
биохимическими сдвигами, нарушениями в системе нейромедиаторов и иммунного
комплекса мозга.
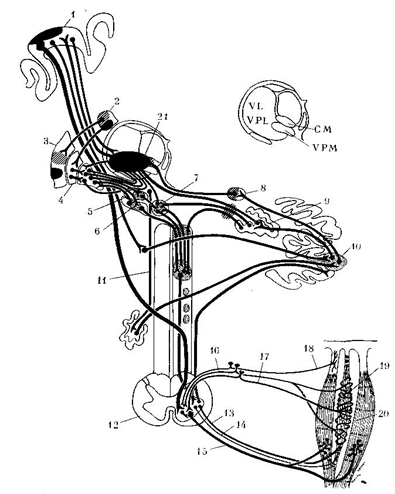
Рис. 1. Схема вовлечения в патологический процесс
при дистониях отдельных образований нервной системы (Соoper,
1965).
1 – моторная кора; 2 – хвостатое ядро; 3 –
скорлупа; 4 – бледный шар; 5 – субталамическое ядро; 6 – черная субстанция; 7 –
красное ядро; 8 – эмболиформное ядро; 9 – зубчатое ядро; 10 – передняя долька
мозжечка; 11 – РФ ствола; 12 – сегмент спинного мозга; 13, 14 –
γ1 и γ2
эфференты; 15 – α-эфферентное
волокно; 16, 17 – афференты I и
II группы в заднем
корешке; 18 – сухожильное тельце Гольджи; 19 – мышечное веретено; 20 –
поперечно-полосатая мышца; 21 – очаг стереотаксической деструкции с наибольшим
терапевтическим эффектом.
Наиболее тяжелыми в клиническом отношении являются, как было
представлено выше, торсионная дистония и спастическая кривошея. Именно поэтому,
вероятно, на сегодняшний день наибольшее число исследований патогенеза относится
к этим двум вариантам дистоний.
Патофизиологические механизмы, лежащие в основе первичной
торсионной дистонии, остаются недостаточно изученными. Большинство новейших
данных по этому вопросу базируется на исследованиях, выполненных у больных с
фокальными дистониями, особенно писчим спазмом. X.O. Breakefield
et al.
(2008) выделяют три вероятных звена патологического процесса:
– распространенное нарушение ингибиторных механизмов, ведущее к
неточностям «фокусировки» и выбора требуемых движений;
– патологическая адаптационная пластичность;
– дезорганизация сенсомоторного процессинга и интеграции.
Например, эффективность сенсорных приемов (корригирующих жестов) в
уменьшении выраженности дистонии позволяет предполагать патофизиологическую роль
нарушений сенсомоторного взаимодействия. Травма периферических нервов -
доказанный фактор, повышающий предрасположенность к дистонии - также может
нарушать нормальные взаимоотношения афферентного и эфферентного потоков в
процессе контроля частых и привычных двигательных актов, вызывая тем самым
сенсомоторный дисбаланс.
До последнего времени при первичной торсионной дистонии не было
описано постоянных нейропатологических изменений. Отсутствие клеточной
дегенерации позволяло говорить о том, что в основе первичной торсионной дистонии
лежат функциональные нарушения соответствующих нейронов. Однако в 2004 г. K.S.
McNaught et
al. описали у 4 больных с
DYTl-дистонией перинуклеарные включения в клетках ретикулярной формации среднего
мозга и околоводопроводного серого вещества, а также тау/убиквитин-иммунореактивные
агрегаты в пигментированных нейронах компактной части черной субстанции и
голубоватого пятна, причем эти изменения отсутствовали в контроле. Последние
работы X.O. Breakefield et
al. (2008), выполненные с
применением специальных режимов нейровизуализации (диффузионно-взвешенной МРТ),
также показывают, что в противоположность ранее существовавшему мнению у
пациентов с фокальной дистонией могут иметь место тонкие анатомические изменения
вещества мозга. Таким образом, высказывается предположение о том, что дистония
является результатом генетической предрасположенности, реализуемой при
триггерном воздействии средовых факторов (патологическое переутомление кисти при
выполнении повторных движений, периферическая травма и др.).
По мнению О.Р. Орловой (1997), динамичность дистонии связана скорее
всего не с определенным анатомическим субстратом, который до сих пор не
обнаружен, а с нарушением взаимодействия между структурами базальных ганглиев,
ствола мозга, таламуса, лимбико-ретикулярного комплекса, двигательной коры
вследствие нарушения обмена нейромедиаторов в этих структурах, что и составляет
органический нейродинамический субстрат дистонии.
Взгляды на патогенетические механизмы развития спастической
кривошеи также до сих пор неоднозначны (Лихачев С.А., Рушкевич Ю.Н., 2005). Л.С.
Петелин (1970) рассматривал данную патологию как высвобожденную стволовую
тоническую реакцию в результате дисфункции оральных отделов ствола. Некоторые
авторы высказывали предположения о влиянии лабиринтопатий и вестибулярных
расстройств ввиду тесных взаимоотношений между вестибулярными структурами и
аппаратом регуляции положения головы и шеи. Указывая на наличие у 2/3 больных
спастической кривошеей вестибулярных нарушений при отсутствие другой
отоневрологической симптоматики либо спонтанного нистагма, направленного в
сторону противоположную насильственному наклону или повороту головы, лабиринтную
дисфункцию. Также было показано, что поражение мозжечково-талламических связей,
принимающих участие в контроле положения головы и шеи, может приводить к
возникновению спастической кривошеи (Кандель Э.И., 1981; Лис А.Д., 1989;
Overview A.,
2000). Подчеркивается важность изменений динамической межполушарной асимметрии,
которые способствуют нарушениям функционального состояния полушарий головного
мозга, срединных неспецифических структур ствола, нейро-мышечного аппарата шеи.
Одна из теорий патогенеза рассматривает роль измененной супрануклеарной
регуляции мотонейронов (Орлова О.Р., 1989). Наибольшее число исследователей
развитие спастической кривошеи связывают с нарушениями синтеза и обмена
нейромедиаторов в базальных ганглиях. Часть авторов предполагает наличие
дефицита дофамина избыток ацетилхолина, другие большее значение придают снижению
концентрации норадреналина в постеролатеральных ядрах гипоталамуса, ядре Льюеса,
голубом пятне и повышению в красных ядрах и верхних бугорках четверохолмия (Лис
А.Д., 1989; Орлова О.Р., 1989; Яхно Н.Н. и др., 1995;
Overview A.,
2000). В экспериментах на животных выявлена заинтересованность в развитии
спастической кривошеи ядер таламуса, особенно вентролатерального,
интерстициального ядра Кахала, субталамической области (Лихачев С.А., Рушкевич
Ю.Н., 2005). Клинические патоморфологические исследования проведены у единичных
больных, при этом описывали изменения в мозжечке, хвостатом ядре, бледном шаре,
скорлупе. По заключениям других авторов не отмечено патологических изменений в
клетках базальных ганглиев.
Нейровизуализационными методами выявлены атрофические изменения
головного мозга у пациентов, страдающих спастической кривошеей, однако степень
их выраженности не отличалась от показателей контрольной группы. При проведении
позитронно-эмиссионного томографического исследования метаболизма глюкозы у 16
больных спастической кривошеей не отмечено существенных различий, но выявлен
двусторонний дисбаланс показателей, которые характеризуют метаболизм в таламусе
и базальных ядрах (Stoessl
A.J.
et al.,
1988; Overview
A., 2000). Исследовались параметры
протонной МР - спектроскопии (SPECT) у 9 пациентов со спастической кривошеей и
13 здоровых лиц. В ходе исследования выявлено значительное снижение отношений
N-ацетил-аспартата к холинсодержащим веществам и креатину по сравнению с
контрольной группой (Moller
H.E.
et al.,
1998; Overview
A., 2000;
Federico F.
et al.,
2001).
Особо следует остановиться на механизмах наследования отдельных
клинических вариантов дистоний. В этом направлении на сегодняшний день
достигнуты, пожалуй, наибольшие успехи, о которых хотелось бы сообщить.
Картированы 4 хромосомных локуса первичных дистоний и выделен
белковый патологический продукт (торсин А) для одного из них (DYT-1).
Доказано, что «классическая» торзионная дистония Оппенгейма (DYT-1)
наследуется по аутосомно-доминантному принципу с пенетрантностью до 30%.
Аутосомно-доминантный тип наследования характерен для болезни Гентингтона,
спиноцеребеллярных атрофий 2, 3, 6-го и 17-го субтипов, болезни Фара (семейная
кальцификация базальных ганглиев), нейроферритинопатии, фронтотемпоральной
деменции. Также выделена группа дистоний, связанных с заболеваниями, имеющими
Х-сцепленный тип наследования (5 нозологий) и митохондриальные болезни (4
заболевания). В большинстве случаев патологические движения обусловлены
дисфункцией базальных ганглиев в комбинации с нарушением синтеза дофамина.
Нейровизуализационные исследования, как правило, выявляют наличие структурной
церебральной патологии.
Развитие аутосомно-доминантной ДОФА-чувствительной дистонии
обусловлено мутациями гена GTP-циклогидролазы I - первого и скорость-
лимитирующего фермента биосинтеза тетрагидробиоптерина. Недостаток последнего
приводит к снижению активности тирозингидроксилазы и дефициту дофамина в
нигростриатных дофаминергических нейронах (Ichinose
H. et
al., 1995).
При миоклонус-дистонии пациенты с семейным анамнезом заболевания
(форма DYT-11) имеют в основном аутосомно-доминантный тип наследования и
получают мутантный ген от отца (Zimprich A. et al., 2001). Особенностью
патологии является хорошее купирование симптомов при приеме алкоголя, поэтому
количество случаев алкогольной зависимости больше у больных с этой патологией,
чем у членов той же семьи без патологии (Saunders-Pullman R. et al., 2002).
Аутосомно-доминантный тип наследования характерен для
дистонии-паркинсонизма; здесь патологический белковый продукт - Na+/K+-АТФаза-α3
- каталитический белок натрий-калиевого насоса (De Carvalho Aguiar P. et al.,
2004).
Важной аутосомно-рецессивной патологией, протекающей с дистонией,
является ювенильный паркинсонизм, обусловленный мутациями гена паркина. При этом
начало заболевания обычно отмечают в возрасте до 40 лет, вначале оно
ассоциируется с дистонией, гиперрефлексией, медленным прогрессированием и ранним
развитием дискинезий, связанных с приемом леводопы. Выраженность дистонии
проявляется ярче в нижних конечностях, хотя отмечают и поражение рук, шеи и
туловища.
Интереснейшие данные о молекулярной генетике наследственных
дистонических синдромов приведены в известной статье С.Н. Иллариошкина и др.
(2000), опубликованной в Журнале неврологии и психиатрии Им. С.С. Корсакова. На
самых интересных сведениях, представленных в данной статье мы позволим себе
подробно остановиться ниже.
Авторы со ссылками на
многочисленные литературные источники пишут о том, что торсионная дистония может
передаваться по аутосомно-доминантному, аутосомно-рецессивному и Х-сцепленному
типам, однако в большинстве случаев наблюдается классический
аутосомно-доминантный путь наследования, как и сообщалось выше. Существует также
группа редких наследственных дистонических синдромов с более избирательной
локализацией и особыми характеристиками гиперкинезов (например,
пароксизмальность их возникновения, фокальный характер дистонии и др.).
Указывается также, что детальный анализ клинического полиморфизма торсионной
дистонии позволил разделить это заболевание на две основные формы: ригидную
и гиперкинетическую (Маркова Е.Д., 1975, 1983, 1989).
Ригидная форма торсионной дистонии, начинающаяся обычно в детском,
реже юношеском возрасте, характеризуется постоянным повышением мышечного тонуса
по экстрапирамидному типу и формированием фиксированных патологических поз,
развитием контрактур в поздней стадии болезни. Чрезвычайно характерным является
наличие у больных дневных флюктуаций в моторике - ухудшение состояния к вечеру и
улучшение двигательных функций утром или после дневного сна. Заболевание чаще
наблюдается у женщин (Иванова-Смоленская И.А. и др., 1998;
Segawa M.
et al.,
1976).
Гиперкинетическая форма торсионной дистонии, начинающаяся, как
правило, в первом десятилетии жизни, характеризуется развитием вычурных
генерализованных гиперкинезов конечностей, туловища и шеи, резко усиливающихся
при ходьбе и приводящих к тяжелой инвалидизации больного. Для обозначения этого
заболевания нередко используются другие термины, такие как генерализованная
дистония с ранним началом (earlyonset generelized dystonia), идиопатическая
торсионная дистония (idiopathic torsion dystonia). Как уже указывалось, данная
форма дистонии особенно часто встречается в этнической группе евреев ашкенази -
около 40-50 случаев на 100 000 человек.
Сведения о присутствии двух выделенных групп торсионной дистонии
приведены в связи с тем, что в основе фенотипических различий двух упомянутых
форм заболевания лежат разнонаправленные изменения метаболизма главных
нейротрансмиттеров в головном мозге. При ригидной форме наблюдается снижение
центральной дофаминергической и серотонинергической трансмиссии с одновременным
повышением активности холинергической системы, тогда как для гиперкинетической
формы характерны обратные соотношения (Бархатова В.П., Маркова Е.Д., 1978;
Adolfsson
R. et
al., 1979;
Breakefield
X.O.
et al.,
1981; Le
Witte P.A.
et al.,
1993; Rajput
A.H.
et al.,
1994; Markova
E.D.,
Ivanova-Smolenskaya
I.A.,
1995). Указанные клинико-биохимические различия двух форм торсионной дистонии и
выявленный факт дофаминергического дефицита при ригидной форме явились
теоретической предпосылкой для назначения больным с ригидной формой препарата
L-ДОФА, являющегося предшественником медиатора дофамина. Это дало основание T.G.
Nygaard et
al. (1988) обозначить данную
клиническую форму ДОФА-зависимой, или ДОФА-чувствительной дистонией (dopa-responsive
dystonia). Напротив, при гиперкинетической форме торсионной дистонии L-ДОФА не
только не улучшает состояния, но даже приводит к усилению гиперкинезов.
T.G. Nygaard et al. (1993) картировали ген
аутосомно-доминантной ДОФА-зависимой дистонии на длинном плече 14-й хромосомы в
нескольких семьях. Вскоре после этого локализация мутантного гена на хромосоме
14q была подтверждена японскими исследователями, что позволило на молекулярном
уровне установить нозологическую идентичность понятий «наследственная
прогрессирующая дистония» и «ДОФА-зависимая дистония» (Tanaka
H. et
al., 1995). В 1994 г. группа
исследователей (Ichinose H.
et al.,
1994), изучавших один из ключевых ферментов дофаминового обмена -
ГТФ-циклогидролазу 1 (ГЦГ-1), идентифицировали ген данного фермента и установили
его локализацию на 14-й хромосоме в интервале 14q21-22, то есть в локусе
ДОФА-зависимой дистонии. Это послужило основанием для исследования гена ГЦГ-1 в
качестве кандидата у больных с аутосомно-доминантной ДОФА-зависимой дистонией.
Были получены прямые доказательства того, что ген ГЦГ-1 ответственен за
возникновение аутосомно-доминантной ДОФА-зависимой дистонии, поскольку
гетерозиготные мутации в нем были обнаружены у больных в 4-х обследованных
семьях. Мутации гена ГЦГ-1 в каждой семье были различными: в 3-х случаях это
были точковые мутации, приводящие к замене кодируемой аминокислоты, а в одном
случае выявлена
делеция со сдвигом рамки считывания и
обрывом
трансляции.
В свете имеющихся данных патогенез аутосомно-доминантной
ДОФА-зависимой дистонии на молекулярном уровне схематично представлен на
рисунке 2. Фермент ГЦГ-1 регулирует
превращение ГТФ в дигидронеоптерин-трифосфат; последний в свою очередь является
субстратом для образования тетрагидробиоптерина (ВН4) -
кофактора
тирозингидроксилазы, ответственной за
синтез дофамина из
тирозина. При наличии мутаций в гене
ГЦГ-1 нарушаются функция данного фермента и весь последующий биохимический
каскад, что приводит к недостаточности дофамина. Синтез дофамина может быть
эффективно восстановлен при введении его предшественника - L-ДОФА, что и имеет
место у больных.

Рис. 2. Патогенез аутосомно-доминантной ДОФА-зависимой дистонии
на молекулярном уровне К настоящему времени идентифицировано свыше 40 различных
(преимущественно точковых) мутаций в кодирующей области гена ГЦГ-1 у больных в
японской, корейской, испанской, британской, немецкой и российской популяциях.
Практически все авторы отмечают, что повреждение гена ГЦГ-1 обусловливает лишь
половину всех случаев с типичной клинической картиной ДОФА-зависимой дистонии.
Это может быть объяснено либо истинной генетической гетерогенностью
аутосомно-доминантной ДОФА-зависимой дистонии, либо повреждениями в некодирующих
областях гена ГЦГ-1 (промоторная зона,
интронные мутации с нарушениями
сплайсинга и др.).
Как уже сказано, у всех больных с аутосомно-доминантной
ДОФА-зависимой дистонией имеется мутация гена ГЦГ-1 в гетерозиготном состоянии.
Следовательно, вторая копия гена на интактной хромосоме у них сохранна, что,
однако, не предотвращает развития болезни. Молекулярный механизм этого явления
был раскрыт M. Hirano et
al. (1998). Авторы показали, что
гетерозиготные мутации в гене ГЦГ-1 характеризуются «доминантным негативным
эффектом», который заключается в том, что наличие мутации на одной из парных
хромосом приводит не только к синтезу мутантной субпопуляции молекул ГЦГ-1, но и
к нарушению функциональной активности нормальной молекулы белка, синтезированной
с неповрежденной копии гена. По-видимому, это объясняется мультимерной
структурой белкового комплекса ГЦГ-1 (состоящего из 10 идентичных субъединиц) и
возможностью взаимодействия нормальных и мутантных молекул ГЦГ-1 (Nar
H. et
al., 1995). Встраивание в
состав данного комплекса даже одной мутантной белковой молекулы приводит к
образованию функционально дефектных гетеромеров, нарушению процессинга белка
и/или преждевременной деградации фермента. Конечным результатом является
патогенетически значимое снижение функции ГЦГ-1.
Интересно отметить (Иллариошкин С.А. и др., 2000), что наличие
мутаций в гене ГЦГ-1 в гомозиготном состоянии (то есть повреждение обеих копий
гена) ведет к манифестации совершенно особого заболевания - атипичной
неонатальной гиперфенилаланинемии (Furukawa
Y.,
Kish S.J.,
1999). Оно обусловлено полным блоком активности тирозингид-роксилазы, глубоким
сочетанным дефицитом катехоламинов - дофамина, норадреналина,
адреналина, серотонина, а также
повышением содержания в крови фенилаланина (последний показатель связан с тем,
что при выраженной недостаточности ВН4 страдает не только
тирозингидроксилаза, но и близкая к
ней фенилаланингидроксилаза). Клинически данный синдром характеризуется
неонатальной задержкой двигательного развития, гипотонией, эпилептическими
припадками, умственной отсталостью. У некоторых больных при особой комбинации
мутантных аллелей гена ГЦГ-1 наблюдается «промежуточный» фенотип болезни с
задержкой психомоторного развития и речи, прогрессирующей дистонией и умеренным
положительным эффектом при назначении L-ДОФА (Furukawa
Y et
al., 1998) . Было показано, что
при синдроме атипичной гиперфенилаланинемии имеет место практически нулевая
активность ГЦГ-1, при «промежуточной» форме активность ГЦГ-1 обычно не превышает
1-2%, у больных ДОФА-зависимой дистонией она составляет 5-20%, а при активности
фермента выше 30-40% (что часто наблюдается у гетерозиготных носителей мутации)
какие-либо клинические проявления недостаточности ГЦГ-1 отсутствуют. Таким
образом, заболевания, обусловленные патологией гена ГЦГ-1, представляют особый
континуум взаимно перекрывающихся состояний - от «стертых» субклинических форм
ДОФА-зависимой дистонии на одном конце спектра до тяжелой неонатальной
гиперфенилаланинемии на другом; при этом степень тяжести клинического синдрома
увеличивается по мере нарастания дозы мутантного гена и дефицита активности
фермента ГЦГ-1.
Иначе обстоит дело с молекулярной генетикой ДОФА-независимой
дистонии. Ген ДОФА-независимой дистонии, обозначаемый DYT1, был картирован L.
Ozelius et
al. (1989). на длинном плече 9-й
хромосомы (локус 9q32-34). Данная локализация мутантного гена была подтверждена
в большинстве семей в разных этнических группах мира. Анализ серии
высокополиморфных маркеров, сцепленных с геном DYT1, показал, что в популяции
евреев ашкенази, характеризующейся высокой частотой ДОФА-независимой дистонии, у
больных отмечается один и тот же характерный гаплотип. Это свидетельствует об
«эффекте основателя» и едином генетическом происхождении ДОФА-независимой
дистонии в данной этнической группе независимо от дальнейшего географического
расселения потомков (Risch
N.J.
et al.,
1995).
Было установлено, что ген DYT1 кодирует неизвестный ранее белок -
торсин А (Ozelius
L.J.
et al.,
1997). Этот белок относится к группе торсинов, точные функции которых у высших
организмов окончательно не выяснены. Торсин А входит в состав олигомерных
белковых комплексов и имеет определенную гомологию с семейством белков
«теплового шока», играющих роль в предохранении клеток от термического стресса.
Эти белки обладают АТФ-связывающей активностью и, помимо регуляции
термотолерантности, могут участвовать в процессах образования везикул,
конформационных изменениях белков, регуляции клеточных сигналов и
функционировании митохондрий. S. Augood et
al. (1998) показали, что торсин
А экспрессируется в дофаминергических нейронах компактной части черной
субстанции, а также в мозжечке и гиппокампе. В совокупности вышеуказанные данные
свидетельствуют о возможной роли торсина А в процессах высвобождения
нейромедиаторов в нигростриарной системе мозга и/или поддержании энергетического
потенциала дофаминергических нейронов и нейронопротекции.
Было обнаружено, что более чем у 70% больных ДОФА-независимой
дистонией из Европы и Северной Америки имеет место один и тот же первичный
дефект - делеция 3
нуклеотидов (гуанин - аденин -
гуанин, GAG) в кодирующей области гена (Ozelius
L.J.
et al.,
1997). Эта делеция ведет к утрате аминокислоты глутамата в карбоксильной части
гена. Данная мажорная мутация обусловливает практически все случаи
ДОФА-независимой дистонии в популяции евреев ашкенази, однако не ограничена
только этой этнической группой и встречается с достаточно высокой частотой у
больных других национальностей (Иллариошкин С.Н. и др., 2000). Если в этнической
группе евреев ашкенази преобладающее носительство делеции GAG у больных
обусловлено «эффектом основателя» и наследованием генетически общей предковой
мутантной хромосомы, то в других этнических группах вышеуказанная делеция
возникает повторно в результате многократных независимых мутационных событий.
Частое возникновение одной и той же мутации у разных больных является
чрезвычайно редким феноменом в клинической генетике.
Анализ клинико-генетических корреляций показал, что универсальная
делеция GAG в гене DYT1 обнаруживается не только у больных с типичной
генерализованной формой ДОФА-независимой дистонии. В некоторых семейных случаях
данная мутация была выявлена также у больных с фокальными (писчий спазм),
мультифокальными и сегментарными формами ДОФА-нечувствительной дистонии, а также
у больных с некоторыми атипичными клиническими проявлениями болезни (постуральный
тремор рук, заикание вследствие дистонии оральной мускулатуры и т.п.). Данный
факт позволяет существенно расширить спектр клинических проявлений 9q-сцепленной
формы наследственной дистонии и должен приниматься во внимание при
медико-генетическом консультировании. В ряде случаев делеция GAG в изучаемой
области гена DYT1 была выявлена также у больных ДОФА-независимой дистонией, не
имевших семейного анамнеза (Ozelius
L.J.
et al.,
1997); большинство указанных случаев, по-видимому, являются «псевдоспорадическими»
и объясняются неполной пенетрантностью мутантного гена в семье либо отсутствием
достоверной генеалогической информации.
Таким образом, с практической точки зрения важно отметить, что
обнаружение универсальной делении GAG в гене DYT1 у значительной части больных
ДОФА-независимой дистонией дает возможность проведения простой и достоверной
ДНК-диагностики в большинстве семейных и части спорадических случаев данной
клинической формы дистонии, а также у клинически здоровых родственников из групп
риска в консультируемых отягощенных семьях.
Помимо рассмотренных основных типов торсионной дистонии, существует
ряд более редких форм наследственной дистонии, которые с
молекулярно-генетической точки зрения охарактеризованы пока в меньшей степени. К
таким формам относится так называемая пароксизмальная дистония, описанная выше.
Одним из вариантов пароксизмальной дистонии является
пароксизмальный дистонический
хореоатетоз (пароксизмальная
некинезигенная дискинезия). Это заболевание имеет аутосомно-доминантный тип
наследования и проявляется приступами сложных непроизвольных движений (дистония,
хореоатетоз), возникающих в покое. В типичных случаях приступ начинается как
гемидистония, но постепенно гиперкинез охватывает все конечности, туловище, шею
и речевую мускулатуру; сознание в момент приступа остается сохранным.
Гиперкинезы длятся от нескольких минут до нескольких часов, нередко
сопровождаются аурой (парестезии) и могут быть спровоцированы приемом алкоголя
или кофеина. Частота двигательных пароксизмов обычно снижается с возрастом. Ген
данного заболевания был картирован на хромосоме 2q33-35 (Fink
J.K.
et al.,
1996; Fouad
G.T.
et al.,
1996).
Осложненной формой пароксизмального дистонического хореоатетоза
является другой редкий наследственный синдром - так называемый пароксизмальный
хореоатетоз со спастичностью. Данное заболевание наследуется по
аутосомно-доминантному типу и проявляется приступами дистонически-хореоатетозных
гиперкинезов продолжительностью несколько минут, обычно провоцируемых физической
нагрузкой; у части больных развивается постоянный (иногда прогрессирующий)
нижний спастический парапарез. Ген пароксизмального хореоатетоза со
спастичностью локализован на хромосоме 1р, в тесной близости от кластера генов
калиевого канала (Auburger
G.et
al., 1996). Предположительно
данный синдром обусловлен мутацией одного из генов указанного кластера, то есть
он может относиться к растущей группе «каналопатий» - пароксизмальных
заболеваний, связанных с повреждением генов калиевых, натриевых и кальциевых
каналов, регулирующих проницаемость и возбудимость клеточных мембран, процессы
секреции и передачи клеточных сигналов.
Особую разновидность пароксизмальных дистоний представляет
пароксизмальный кинезигенный хореоатетоз, отличающийся тем, что приступы
дискинезии у больных тесно связаны с внезапными движениями конечностей (чаще
ног), например при беге или резком вставании с кресла. Провоцирующими факторами
могут служить стресс, действие неожиданных сенсорных стимулов, усталость, холод,
менструация. Длительность двигательных пароксизмов мала (от 3 до 40 сек.),
нередко приступу предшествует аура в виде парестезии или мышечного напряжения,
сознание не нарушается. Заболевание наследуется по аутосомно-доминантному типу.
F. Picard et
al. (1998) показали достоверное
отсутствие сцепления пароксизмального кинезигенного хореоатетоза с локусами
описанных выше форм пароксизмальных дистоний на хромосомах 2q и 1р, что
свидетельствует о нозологической самостоятельности данного заболевания.
Наконец, к настоящему времени установлена хромосомная локализация
генов еще трех редких вариантов наследственной дистонии. Это «смешанная» форма
наследственной дистонии; фокальная дистония с поздним началом и Х-сцепленный
синдром «дистония-паркинсонизм» (синдром Lubag).
Тип наследования «смешанной» формы наследственной дистонии -
аутосомно-доминантный, отмечается неполная пенетрантность мутантного гена.
Дистония манифестирует в первом - втором десятилетии жизни и нечувствительна к
препаратам L-ДОФА, что сближает ее с классической DYT1-ассоциированной
ДОФА-независимой дистонией с ранним началом. Однако в отличие от DYT1-дистонии
при «смешанной» форме наследственной дистонии у большинства больных развиваются
фокальные формы заболевания с преимущественным вовлечением краниоцервикальной
мускулатуры (шея, лицо, артикуляционная мускулатура), тогда как генерализация
процесса имеет место лишь у небольшого числа больных. L. Almasy
et al..
(1997) при исследовании 2-х семей с данной формой дистонии локализовали ген
заболевания на хромосоме 8p21-q22. Данный хромосомный локус обозначен DYT6.
2. Фокальные дистонии с поздним началом (спастическая кривошея,
писчий спазм, спастическая дисфония и т.п.) представляют собой самую
распространенную форму идиопатической дистонии. Есть основания предполагать
определенную poль в их развитии генетических факторов, поскольку описаны
единичные семьи с аутосомно-доминантным типом передачи различных фокальных форм
дистонии. В одной из таких больших семей, в которой у ряда родственников из двух
поколений в возрасте от 28 до 70 лет манифестировали симптомы фокальной дистонии
разной локализации (кривошея, спастическая дисфония,
оромандибулярная дистония, писчий
спазм), B. Leube et
al. (1997) картировали ген заболевания
на хромосоме 18p. B современной номенклатуре данный хромосомный локус имеет
обозначение DYT7. Результаты исследований оказались, тем не менее,
противоречивыми и не позволяют пока с достоверностью судить о роли локуса DYT7 в
этиологии спорадических фокальных дистоний.
Х-сцепленный синдром «дистония-паркинсонизм» является
популяционно-специфичным и наблюдается главным образом у аборигенов Филиппинских
островов (Иллариошкин С.Н. и др., 2000). Тип наследования ясен из названия.
Заболевание начинается во втором - четвертом десятилетии жизни с фокальных
дистонических гиперкинезов лица, оромандибулярной мускулатуры, шеи, туловища,
конечностей с постепенным развитием генерализованной формы дистонии. По мере
прогрессирования болезни постепенно присоединяется синдром паркинсонизма,
нечувствительный к препаратам L-ДОФА. Патологический ген (DYT3) локализован в
перицентромерной области Х-хромосомы, в локусе Xql3.1 (Graeber
M.B
et al.,
1992; Haberhausen
G et
al., 1995).
Многочисленные данные подтверждают выраженную генетическую
гетерогенность наследственной дистонии и свидетельствуют о наличии ряда других,
пока не идентифицированных генов, повреждение которых лежит в основе развития
наследственных дистонических синдромов. Успехи молекулярной генетики приводят к
постепенному внедрению в классификацию наследственных дистоний геномного
принципа, что является общей закономерностью для всех наследственных заболеваний
нервной системы. Исследование группы наследственных дистоний с использованием
методов молекулярной медицины открывает перспективы разработки и внедрения
высокоэффективных патогенетических и этиологических методов лечения этих тяжелых
страданий (Иллариошкин С.Н. и др., 2000). |