Данная информация предназначена для специалистов в области здравоохранения и фармацевтики. Пациенты не должны использовать эту информацию в качестве медицинских советов или рекомендаций.
Современные подходы к диагностике наследственных нарушений
соединительной ткани
А. В. Клеменов, доктор медицинских наук
А. С. Суслов
ГБУЗ НО ГКБ № 30, Нижний Новгород
Наследственные нарушения соединительной ткани (ННСТ) или, как их еще
называют в России, дисплазии соединительной ткани — одна из наиболее
дискуссионных проблем клинической медицины. До недавних пор в нашей стране
существовала терминологическая путаница и отсутствие единого подхода к оценке
этих состояний. Главным образом, это касалось так называемых
недифференцированных ННСТ, включавших все варианты врожденной «слабости»
соединительной ткани за исключением моногенных синдромов Марфана, Элерса–Данло и
ряда других. Отсутствие четких диагностических критериев приводило к тому, что
любые случаи выявления каких-либо признаков дизэмбриогенеза произвольно
обозначались как ННСТ [1]. Подобная широкая и необоснованная трактовка приводила
к гипердиагностике, создавала предпосылки для психогенных ятрогений.
Для преодоления существующих противоречий в дефинициях и критериях диагноза
отдельных клинических вариантов ННСТ комитет экспертов Всероссийского научного
общества кардиологов (ВНОК) разработал первые национальные рекомендации,
принятые на Российском национальном конгрессе кардиологов в 2009 г. и
пересмотренные в 2012 г. [2]. Эти усилия позволили существенно сблизить подходы
к диагностике ННСТ в нашей стране с международной практикой.
Термин «ННСТ» объединяет генетически и клинически гетерогенную группу
заболеваний на основе общности нарушений формирования соединительной ткани в
эмбриональном и постнатальном периодах. Генетическая гетерогенность ННСТ
подразумевает моногенную и мультифакториальную природу заболевания. Первая
реализована в группе относительно редких моногенных синдромов Марфана и Элерса–Данло,
сопряженных с мутациями генов белков внеклеточного матрикса. В возникновении
наиболее многочисленной группы ННСТ мультифакториальной природы значимы как
мутации большого числа различных генов, так и воздействие факторов внешней
среды. Клиническая гетерогенность ННСТ связана с повсеместным распространением в
организме соединительной ткани и многообразием проявлений врожденной «слабости»
ее отдельных компонентов.
Поскольку для большинства ННСТ отсутствуют специфические лабораторные
маркеры, а молекулярно-генетические исследования остаются малодоступными и
значимыми лишь по отношению к моногенным вариантам патологии, приоритет в
диагностике остается за клиническими признаками. В упомянутых выше рекомендациях
подобные признаки систематизированы, из них выделены те, которые имеют
наибольшее диагностическое значение и включены в опубликованные зарубежные
рекомендации по диагностике наиболее изученных ННСТ (Гентские критерии синдрома
Марфана [3], Вилльфраншские критерии синдрома Элерса–Данло [4], Брайтонские
критерии синдрома гипермобильности суставов [5]). Существенно, что от этих
признаков четко отграничены стигмы дисэмбриогенеза (малые аномалии развития),
которые хотя и выявляются при ННСТ чаще, чем в общей популяции (что подтверждает
роль нарушений эмбриогенеза в формировании ННСТ), но собственно маркерами
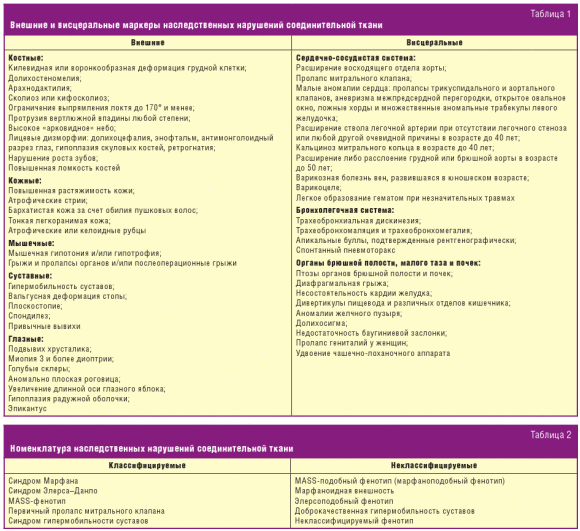
«слабости» соединительной ткани не являются. Перечень основных внешних и
висцеральных маркеров ННСТ приведен в табл. 1. Совокупность выявленных признаков
у конкретного пациента позволяет диагностировать тот или иной вариант
соединительнотканной патологии.
В настоящее время применительно к ННСТ рекомендовано отказаться от признанных
устаревшими терминов «дифференцированные» и «недифференцированные» и предложено
говорить о нарушениях классифицируемых (имеющих согласованные рекомендации по
диагностике) и неклассифицируемых (или диспластических фенотипах) — табл. 2 [1,
2]. Согласованные рекомендации по диагностике имеют: из моногенных ННСТ —
синдромы Марфана и Элерса–Данло, из мультифакториальных — MASS-фенотип,
первичный пролапс митрального клапана, синдром гипермобильности суставов.
Синдром Марфана — аутосомно-доминантная патология, в основе которой лежит
мутации гена фибриллина-1 (FBN1). Фибриллин составляет основу эластических
волокон; его особенно много в межклеточном матриксе сосудистой стенки, сердца,
хрящей, хрусталика, роговицы и цинновой связки. Мутации гена FBN1 приводят к
неполноценности фибриллина и нарушению структуры и функции перечисленных органов
и тканей.
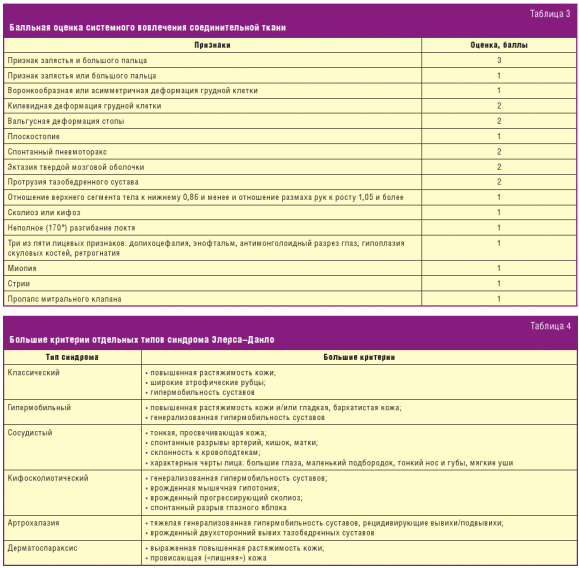
Диагностика синдрома Марфана основана на Гентских критериях (1996, 2010 гг.).
В последней версии Гентских критериев [3] было упразднено деление на большие и
малые признаки, ряд малых признаков исключен. Одновременно было выделено два
наиболее специфичных признака — дилатация и/или расслоение аорты и эктопия
хрусталика и предложена балльная оценка остальных признаков для расчета степени
системного вовлечения соединительной ткани (СВСТ) — табл. 3. В отсутствие
семейного анамнеза диагноз синдрома Марфана может быть установлен при наличии
расширения корня аорты и эктопии хрусталика либо при сочетании расширения аорты
с мутацией гена FBN1 или с совокупностью признаков СВСТ на 7 и более баллов. При
отягощенном семейном анамнезе диагноз правомерен, если выявляется один из
специфичных признаков или если СВСТ составляет 7 и более баллов.
Синдром Элерса–Данло — гетерогенная группа коллагенопатий с различными типами
наследования и общими клиническими проявлениями в виде гипермобильности суставов
и повышенной эластичности кожи. Диагностика синдрома Элерса–Данло основана на
Вильфраншских критериях [4]. Вместо ранее признаваемых десяти типов болезни в
настоящее время выделены шесть: классический, гипермобильный, сосудистый,
кифосколиотический, артрохалазия, дерматоспараксис; для каждого из них
определены большие и малые диагностические критерии. Для клинической диагностики
необходимо наличие хотя бы одного большого критерия (табл. 4).
MASS-фенотип (или марфаноподобный синдром) — акроним, обозначающий пролапс
митрального клапана (Mitral valve prolapse), расширение аорты (Aotic dilatation),
изменения кожи (Skin) и костей скелета (Skeleton). MASS-фенотип можно
диагностировать при пограничном расширении корня аорты, наличии хотя бы одного
скелетного проявления и признаков СВСТ на 5 и более баллов. Как можно заметить,
при отсутствии данных молекулярно-генетической диагностики MASS-фенотип трудно
(если вообще возможно) отличить от синдрома Марфана с неполным набором
признаков.
Пролапс митрального клапана диагностируется при систолическом смещении одной
или обеих створок митрального клапана за линию клапанного кольца в
парастернальной продольной позиции более чем на 2 мм. Морфологическим субстратом
первичного пролапса митрального клапана как одного из вариантов ННСТ выступает
миксоматоз створок, отражающий дезорганизацию коллагеновых фибрилл и накопление
в них кислых гликозаминогликанов.
При оценке пролапса митрального клапана рекомендуется обращать внимание на
глубину пролабирования, толщину створок и степень митральной регургитации — эти
параметры существенны для прогнозирования нарушений внутрисердечной и общей
гемодинамики. При высокой степени митральной регургитации и толщине створки
более 5 мм (признак ее миксоматозной дегенерации) вероятность гемодинамических
расстройств достоверно повышается. Придается значение и признакам СВСТ как
весомому подтверждению принадлежности пролапса к ННСТ (кроме первичного
существуют и вторичные пролапсы митрального клапана, не связанные с врожденной
«слабостью» соединительной ткани, а развивающие при поражениях миокарда левого
желудочка — миокардитах, миокардиодистрофии, коронарной патологии). Если
пролабирование створок митрального клапана составляет не более 2 мм, они не
утолщены, а митральная регургитация отсутствует или минимальна, оснований
констатировать патологию нет. В этом случае может идти речь о варианте нормы у
лиц астенической конституции или преходящем «физиологическом» пролапсе у
подростков.
Первичный пролапс митрального клапана следует отличать от митрального
пролапса как принадлежности моногенных ННСТ или MASS-фенотипа. Дифференциальными
критериями (к сожалению, не абсолютными) являются диаметр аорты и количество
признаков СВСТ.
В основе синдрома гипермобильности суставов лежат мутации генов, кодирующих
коллаген, эластин, фибриллин и тенасцин Х, приводящие к слабости суставных
связок. Синдром характеризуется избыточным диапазоном движений в суставах,
сопровождающимся клинической симптоматикой (привычные вывихи, артралгии). При
диагностике гипермобильности суставов используется девятибалльная шкала P.
Beighton [5], предусматривающая оценку способности выполнения следующих пяти
движений: пассивного сгибания V пястно-фалангового сустава более чем на 90°,
пассивного приведения I пальца к предплечью, пассивного переразгибания коленных
и локтевых суставов более 10°, свободного касания ладонями пола при прямых
ногах. Первые четыре движения — парные (присваивается по баллу за возможность
выполнить движение на каждой из сторон), последнее — непарное (максимально
возможный суставной счет — 9 баллов). Гипермобильность суставов, составляющая не
менее 4 баллов, и артралгии не менее чем в четырех суставах продолжительностью
от трех месяцев и являются большими диагностическими критериями данной
патологии.
Поскольку слабость связочного аппарата является универсальным признаком
соединительнотканной недостаточности, синдром гипермобильности суставов
исключается при наличии синдромов Марфана, Элерса–Данло и ряда других близких им
по клиническим проявлениям ННСТ.
Неклассифицируемые ННСТ, не подходящие под согласованные критерии
диагностики, встречаются в повседневной практике гораздо чаще. Многообразие их
клинических вариантов систематизировано в следующие варианты: МASS-подобный
фенотип, марфаноидная внешность, элерсоподобный фенотип, доброкачественная
гипермобильность суставов, неклассифицируемый фенотип. Первые два из них
фенотипически напоминают синдром Марфана, два следующие — синдром Элерса–Данло,
не отвечая полностью критериям диагноза указанных состояний. В основу
диагностики неклассифицируемых ННСТ положены те же принципы (совокупность
внешних и висцеральных фенотипических проявлений), что используются при
выявлении ННСТ, имеющих согласованные рекомендации, однако диагностический порог
при этом менее высокий.
MASS-подобный (марфаноподобный) фенотип характеризуется пограничным значением
размера корня аорты в сочетании с миопией и/или пролапсом митрального клапана и
наличием признаков СВСТ менее 5 баллов (в отличие от MASS-фенотипа, при котором
— 5 баллов и более).
Марфаноидная внешность характеризуется только признаками вовлечения костной
системы (обычно у астеников) при отсутствии висцеральных изменений. При этом
допускаются менее строгие скелетные изменения, чем те, что необходимы для
констатации синдрома Марфана, однако наличие долихостеномелии и арахнодактилии
признается обязательным.
Главное условие отнесения пациента к элерсоподобному фенотипу — наличие не
менее двух признаков вовлечения кожи, исключая большие критерии синдрома Элерса–Данло.
Доброкачественная гипермобильность суставов констатируется на основе
выявления избыточного диапазона движений в суставах, но без клинической
симптоматики.
К неклассифицируемому фенотипу предложено относить случаи выявления не менее
шести малых внешних и/или висцеральных признаков врожденной «слабости»
соединительной ткани, не попадающие под критерии других вышеназванных синдромов
и фенотипов.
Неспецифичность внешних и висцеральных маркеров «слабости» соединительной
ткани, известная условность диагностических критериев диспластических фенотипов
(некоторые из которых отличаются не качественно, а количественно — по числу
констатированных признаков) затрудняют распознавание отдельных ННСТ. В процессе
диагностики следует руководствоваться своеобразной иерархией ННСТ, составляющей
непрерывный фенотипический континуум: от моногенных синдромов через
диспластические фенотипы к неклассифицируемому фенотипу и норме. В соответствии
с этим подходом наличие признаков синдрома Марфана или Элерса–Данло исключает
диагноз неклассифицируемой ННСТ. Наличие критериев MASS-фенотипа (в числе
которых фигурируют пролапс митрального клапана и изменения скелета) не дает
оснований говорить о первичном пролапсе митрального клапана или марфаноидной
внешности. Точно так же диагноз первичного пролапса митрального клапана
отвергает заключение о любом из диспластических фенотипов. Наименьший
клинический и диагностический вес имеет неклассифицируемый фенотип.
Литература
- Земцовский Э. В. Недифференцированные дисплазии соединительной
ткани. Попытка нового осмысления концепции // Вестник медицины Северного
Кавказа. 2008; 2: 8–14.
- Наследственные нарушения соединительной ткани в кардиологии. Диагностика и
лечение. Российские рекомендации (I пересмотр) // Российский кардиологический
журнал. 2013; 1 (Прил. 1): 1–32.
- Loeys B. L., Dietz H. C., Braverman A. C. et al. The Revised
Ghent Nosology for the Marfan Syndrome // J. Med. Genetics. 2010; 4: 476–485.
- Beighton P., De Paepe A., Steinmann B. et al. Ehlers-Danlos
syndromes: Revised nosology, Villefranche, 1997 // Am. J. Med. Genetics. 1998;
1: 31–37.
- Grahame R., Bird H. A., Child A. The revised (Brighton, 1998)
criteria for the diagnosis of benign joint hypermobility syndrome // J.
Rheumatology. 2000; 7: 1777–1779.
Статья опубликована в журнале
Лечащий Врач
